

Bài viết Vấn đề kiềm-toan trong toan ceton do đái tháo đường – Tải file PDF Tại đây.
Biên dịch: Bác sĩ Huỳnh Phạm Hoàng Nam
Kamel S. Kamel, M.D., and Mitchell L. Halperin, M.D.
Bài này tập trung vào 3 vấn đề chính của bác sĩ lâm sàng chăm sóc bệnh nhân nhiễm toan ceton do đái tháo đường; tất cà đều liên quan các rối loạn kiềm – toan. Vấn đề đầu tiên là khoảng trống anion huyết tương và tính tỷ lệ thay đổi trong khoảng trống này với sự thay đổi trong nồng độ bicarbonate huyết tương trên những bệnh nhân này; mối quan tâm thứ 2 là truyền tĩnh mạch natri bicarbonate và thứ ba là nhiễm toan nội bào có thể góp phần diễn tiến phù não, đặc biệt ở trẻ em bị nhiễm toan ceton do đái tháo đường. Trong bài báo này, chúng tôi kiểm tra các dữ liệu sẵn có và cố gắng tổng hợp dữ liệu với các nguyên lý sinh học, điều hòa chuyển hóa và cung cấp hướng dẫn lâm sàng
Bicarbonate huyết tương và Khoảng trống anion huyết tương
Sự tích tụ ceton acid trong dịch ngoại bào làm mất các anion bicarbonate và làm tăng các anion ceton acid. Do tăng đường huyết gây bài niệu thẩm thấu và bài tiết natri thẩm thấu, bệnh nhân nhiễm toan ceton do đái tháo đường thường có biểu hiện giảm thể tích dịch ngoại bào rõ rệt. Yếu tố này làm ảnh hưởng đến đánh giá trạng thái cân bằng kiềm toan và một số phương pháp điều trị.
Xác định mức độ nặng của toan chuyển hóa máu thường dựa vào mức độ giảm của nồng độ bicarbonate huyết tương. Tuy nhiên, như trong phương trình dưới đây mô tà, nồng độ bicarbonate huyết tương chỉ giảm vừa phải khi có cà sự thiếu hụt lớn của bicarbonate trong dịch ngoại bào và sự giảm nặng của thể tích dịch ngoại bào. Thiếu hụt bicarbonate là bằng chứng rõ ràng trong quá trình tái mở rộng thể tích dịch ngoại bào khi dùng nước muối sinh lý:
Nồng độ bicarbonate dịch ngoại bào [HCO3–] = Lượng HCO3– trong dịch ngoại bào 4 thể tích dịch ngoại bào.
Người ta thấy bổ sung các anion mới làm tăng khoảng trống anion huyết tương, đây là khác biệt giữa nồng độ các cation chính trong huyết tương (Na+) và các anion chính trong huyết tương (Cl- và HCO3-). Sự khác biệt này chủ yếu do mạng lưới hóa trị anion trong protein huyết tương, chủ yếu là albumin. Một cạm bẫy khi dùng khoảng trống anion huyết tương là không thể hiệu chỉnh được hóa trị âm do albumin huyết tương. Việc điều chỉnh này phải được thực hiện không chỉ khi nồng độ albumin huyết tương giảm mà cà khi nó tăng lên; trường hợp thứ hai có thể xảy ra trên bệnh nhân nhiễm toan ceton do đái tháo đường giảm rõ rệt thể tích dịch ngoại bào. Đối với mỗi lần giảm 1g/dl nồng độ albumin huyết tương với giá trị bình thường là 4g/dl, người ta nên bổ sung 2.5 mmol/l vào giá trị tính toán của khoảng trống anion huyết tương. Với mỗi lần tăng 1g/dl nồng độ albumin huyết tương, người ta nên trừ đi 2.5 mmol/l từ giá trị tính toán của khoảng trống anion huyết tương. Ngay cà với điều chỉnh này, có vẻ như hóa trị âm của albumin đã tăng lên khi có sự giảm đáng kể trong thể tích máu động mạch hiệu quà và do đó tăng giá trị khoảng trống anion huyết tương.
Mối quan hệ giữa tăng (Δ) trong nồng độ bicarbonate huyết tương và giảm (Δ) trong nồng độ bicarbonate huyết tương (Δ-Δ). được dùng để ước tính mức độ tải lượng acid và để phát
hiện sự xuất hiện các rối loạn toan kiềm đồng mắc. Một vài nghiên cứu chứng minh toan ceton do đái tháo đường, tỷ lệ giữa tăng khoảng trống anion huyết tương với giảm nồng độ bicarbonate huyết tương là khoảng 1. Điều quan trọng để nhận ra tỷ lệ này dựa trên “nồng độ” chứ không phải “hàm lượng” và phải tính đến những thay đổi trong thể tích dịch ngoại bào khi sử dụng tỷ lệ này để đánh giá tải lượng acid máu. Ví dụ, hãy xem xét bệnh nhân nữ mắc đái tháo đường típ 1 nặng 50kg và thể tích dịch ngoại bào trong trạng thái ổn định là 10 lít. Bệnh nhân có nồng độ bicarbonate huyết tương là 25 mmol/l và khoảng trống anion huyết tương là 12 mmol/l. Sau khi nhiễm toan ceton do đái tháo đường, bệnh nhân có nồng độ bicarbonate huyết tương giảm xuống còn 10 mmol/l và khoảng trống anion tăng lên 27 mmol/l. Bởi vì tăng đường huyết gây bài niệu thầm thấu và bài tiết natri thầm thấu liên quan nhiễm toan ceton đái tháo đường, thể tích dịch ngoại bào hiện tại chỉ khoảng 7 lít. Mặc dù tỷ lệ giữa thay đổi trong khoảng trống anion huyết tương và thay đổi trong nồng độ bicarbonate huyết tương là 1:1, sự thiếu hụt bicarbonate và lượng ceton acid được giữ lại trong dịch ngoại bào không bằng nhau. Mức giảm hàm lượng các ion bicarbonate huyết tương là 180 mmol ([25 mmol/l x 10 lít] – [10 mmol/l x 7 lít]), trong khi đó mức tăng các anion ceton acid chỉ là 105 mmol ([khoảng 0 mmol/l x 10 lít] + [15 mmol/ l x 7 lít]).
Những tính toán này cho thấy một thành phần khác của bicarbonate mất khi toan ceton (tức là các anion ceton acid được bài tiết trong nước tiểu cùng với các ion Na+ hoặc K+); đây là 1 dạng mất natri bicarbonate gián tiếp (hình 1) mà không biểu hiện bằng tăng khoảng trống anion huyết tương. Một khi thể tích dịch ngoại bào được gia tăng bằng cách truyền nước muối sinh lý, sự thiếu hụt các ion bicarbonate trở thành bằng chứng với các bác sĩ lâm sàng, bởi vì giảm khoảng trống anion huyết tương sẽ không phù hợp với tình trạng tương tự khi tăng nồng độ các ion bicarbonate.
Mất natri bicarbonate gián tiếp này có thể là dạng chiếm ưu thế (và đôi khi là duy nhất) của nhiễm toan chuyển hóa ở một số bệnh nhân nhiễm toan ceton do đái tháo đường.
Điều trị bằng Natri Bicarbonate
Dùng natri bicarbonate để điều trị toan chuyển hóa cấp gây ra do sản xuất quá mức acid hữu cơ đang còn tranh cãi. Toan máu nặng có thể liên quan giảm khả năng co bóp tim, giảm đáp ứng catecholamin nội sinh và có khuynh hướng gây rối loạn nhịp tim, tất cả điều này góp phần làm rối loạn huyết động. Ngoài ra, toan máu nặng có thể cản trở sự gắn kết insulin với thụ thể của nó, dẫn đến giảm khả năng của insulin trong việc làm chậm tốc độ sản sinh toan ceton.
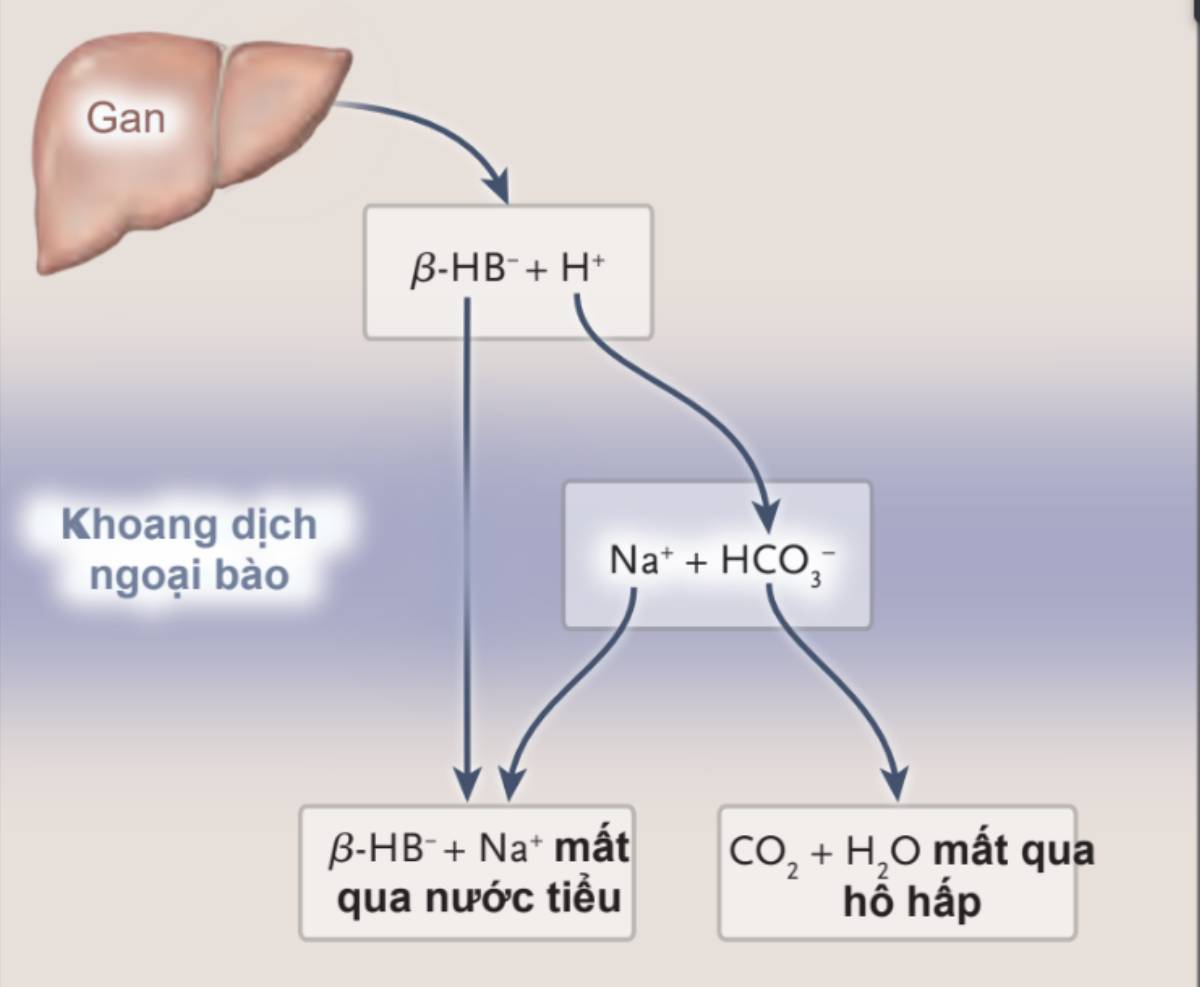
β-hydroxybutyric acid được sản xuất tại gan, phân ly trong dịch ngoại bào thành ^-hydroxybutyrate anion (^-HB–) và hydrogen ion (H+). Mất bicarbonate (HCO3–) và Na+ xảy ra gián tiếp bằng 2 con đường khác nhau. Mất ion bicarbonate xảy ra khi nó phản ứng với H+ tạo ra CO2 và nước (H20), CO2 mất qua quá trình hô hấp. Na+ mất qua bài xuất nước tiểu với ^-hydroxybutyrate.
Trong 3 thử nghiệm ngẫu nhiên có đối chứng trên tổng số 73 bệnh nhân, các nhà nghiên cứu nhận thấy ảnh hưởng của natri bicarbonate ở người trưởng thành với nhiễm toan ceton do đái tháo đường. Bệnh nhân có bệnh đồng mắc nặng (như nhồi máu cơ tim cấp, xuất huyết tiêu hóa, suy thận mạn hoặc nhiễm trừ trong ổ bụng) được loại trừ khỏi 3 thử nghiệm này. Trên cơ sở đánh giá kết quà cơ bàn như thay đổi pH động mạch, nồng độ bicarbonate huyết tương và mức độ các chất chuyển hóa, sử dụng natri bicarbonate không mang lại lợi ích. Áp lực động mạch trung bình chỉ được báo cáo trong 1 nghiên cứu. Chúng tôi không thể tìm thấy những kết quà được công bố từ những thử nghiệm có đối chứng nghiên cứu về ảnh hưởng của natri bicarbinate đối với tỷ lệ tử vong, ổn định huyết động hoặc tỷ lệ mắc các biến chứng như nhồi máu cơ tim, tổn thương thận cấp hoặc đột quỵ trên bệnh nhân toan máu nặng.
Trên bệnh nhân có nồng độ bicarbonate huyết tương rất thấp, bổ sung một lượng nhỏ hydrogen sẽ tạo ra mức giảm tương ứng lớn hơn ở cà nồng độ bicarbonate huyết tương và pH. Ví dụ, giảm 1 nửa nồng độ bicarbonate huyết tương sẽ làm pH động mạch giảm còn 0.3 đơn vị nếu áp suất riêng phần của CO2 trong máu động mạch (PaCO2) không giảm (và trong hầu hết trượng hợp, PaCO2 sẽ không _giảm , do bệnh nhân đang trong tình trạng thông khí tối đa).
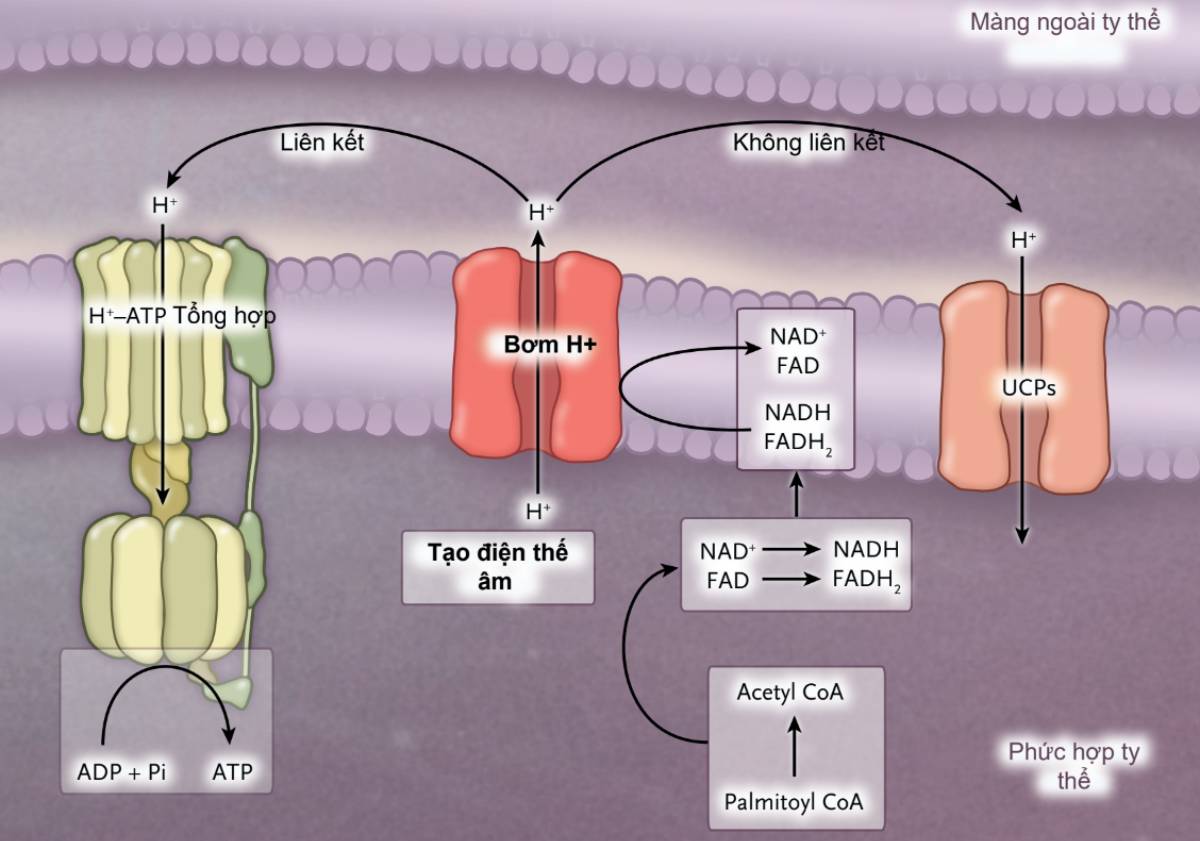
Để dự đoán bệnh nhân nhiễm toan ceton do đái tháo đường có thể bị toan máu nặng hơn, người ta phải hiểu các yếu tố ảnh hưởng tốc độ sàn xuất và loại bỏ toan ceton. Quá trình oxy hóa acid béo chuỗi dài (như palmitic acid) trong ty thể của gan tạo a coenzyme A (tiền chất tạo toan ceton); trong khi đó nicotinamide adenine dinucleotide (NAD+) bị khử thành NADH và flavin adenine dinucleotide (FAD) bị khử thành dạng hydroquinone, FADH2.28 Do các đồng yếu tố quan trọng này chỉ hiện diện với nồng độ rất nhỏ trong ty thể, NADH phải được chuyển thành NAD+ và FADH2 phải được chuyển thành FAD nếu quá trình sàn xuất toan ceton tiếp tục. Sự chuyển đổi này xảy ra trong quá trình phosphoryl hóa oxy hóa liên kết (hình 2), trong đó adenosine triphosphate (ATP) được tái tạo từ adenosine diphosphate (ADP). Ngược lại, quá trình thủy phân ATP (để làm chức năng sinh học) dẫn đến sự hình thành ADP. Do đó, tốc độ thực hiện chức năng sinh học và do sẵn có của ADP đã đặt ra giới hạn về tốc độ phosphoryl hóa oxy hóa. Trên bệnh nhân nhiễm toan ceton do đái tháo đường ăn ít protein, bổ sung amino acid là không đủ để cho phép tăng tốc độ tổng hợp protein ở gan, quá trình này sử dụng ATp.
NADH thành NAD+ (và FADH2 thành FAD), do đó đặt ra giới hạn tốc độ sàn xuất toan ceton.
Tỷ lệ quan sát thấy khi sàn xuất toan ceton trong thời gian đói kéo dài (khoảng 1500 mmol/ngày) cho thấy có nhiều con đường trong gan có thể vượt qua giới hạn sàn xuất toan ceton do cung cấp không đủ ADP. Một khả năng là quá trình phosphoryl hóa oxy hóa không liên kết, trong đó các ion hydro quay lại ty thể bằng các kênh ion hydro tách rời với quá trình chuyển ADP thành ATP (Hình 2).
Trừ khi quá trình phosphoryl hóa oxy hóa tách rời rõ rệt trong quá trình nhiễm toan ceton do đái tháo đường, có khả năng tốc độ sàn sinh toan ceton sẽ không cao hơn đáng kể so với những bệnh nhân nhiễm ceton máu vì nhịn đói kéo dài. Do đó, độ nặng của toan máu có thể diễn tiến nghiêm trọng trên bệnh nhân nhiễm toan ceton do đái tháo đường do giảm chuyển hóa loại bỏ toan ceton.
Toan ceton chủ yếu bị oxy hóa ở não và thận. Các nguyên tắc điều hòa quá trình trao đổi chất tương tự áp dụng cho việc loại bỏ chúng (nghĩa là tốc độ sử dụng ATP đặt làm giới hạn trên cho tốc độ oxy hóa năng lượng, trong trường hợp không có tốc độ thích hợp phosphoryl hóa oxy hóa không liên kết). Bệnh nhân nhiễm ceton do nhịn đói kéo dài chỉ nhiễm toan máu mức độ nhẹ, do tốc độ đào thài toan ceton ở não và thận phù hơp với tốc độ sàn xuất toan ceton ở gan. Trong quá trình nhiễm ceton do nhịn đói kéo dài, não có thể oxy hóa khoảng 800 mmol toan ceton mỗi ngày. Nếu tốc độ tái hấp thu natri ở thận diễn ra bình thường, thận sẽ oxy hóa khoảng 250 mmol toan ceton và bài tiết khoảng 150 mmol các anion toan ceton (phần lớn là amoni NH4+) mỗi ngày. Trong những trường hợp như vậy, cân bằng kiềm toan được bào tồn; sự chuyển hóa các anion toan ceton hoặc bài tiết qua nước tiểu dưới dạng NH4+ là như nhau vì các ion bicarbonate mới được sàn xuất khi các ion NH4+ được bài tiết.
Hầu hết bệnh nhân nhiễm toan ceton do đái tháo đường không cần phải truyền natri bicarbonate, bởi vì insulin truyền vào sẽ làm chậm tốc độ sàn sinh ceton acid và các ion bicarbonate sẽ được tạo ra khi các anion ceton acid bị oxy hóa. Tuy nhiên, sau khi dùng insulin, tốc độ sàn xuất ceton acid có thể giảm trong vài giờ. Ngoài ra, sàn xuất các ionbicarbonate sẽ giảm nếu não và thận oxy hóa ceton acid ít hơn. Tốc độ sử dụng ATP trong não giảm khi hôn mê và khi an thần bằng thuốc hoặc rượu, bởi vì các tình trạng này có thể làm giảm quá trình chuyển hóa trong não. Trên bệnh nhân có độ lọc cầu thận (eGFR) rất thấp do giảm rõ rệt thể tích máu động mạch hiệu quả, việc loại bỏ ceton acid của thận giảm do giảm tốc độ oxy hóa hydroxybutyrate và giảm bài tiết NH4+. Hơn thế nữa, khi tăng thể tích máu động mạch hiệu quả xảy ra khi truyền nhanh nước muối sinh lý có thể dẫn đến giảm thêm nồng độ bicarbonate huyết tương, thứ nhất là do pha loãng và thứ 2 do các ion bicarbonate được loại bỏ do các hydrogen liên kết với các protein nội bào trong cơ. Các ion hydrogen này đươc phóng thích theo mức PaC02 trong máu ở các mao mạch cơ giảm khi dòng máu đến cơ được cải thiện và do đó hệ đệm bicarbonate làm việc hiệu quả hơn để loại bỏ lượng hydrogen. Ngoài ra, khi eGFR tăng, bài tiết nước tiểu các ion ceton acid dẫn đến làm mất các anion có thể được chuyển hóa để sản xuất các ion bicarbonate.
Mặc dù còn nhiều ý kiến trái chiều, không nên truyền natri bicarbonate trên bệnh nhân nhiễm toan ceton do đái tháo đường trừ khi pH động mạch <6.9, chúng tôi đề xuất quyết định điều trị này trên bệnh nhân trưởng thành nhiễm toan ceton do đái tháo đường phải cá thể hóa điều trị và không nên chỉ dựa vào giá trị pH máu.
Điều trị bằng natri bicarbonate có yêu cầu trên bệnh nhân mà 1 thành phần lớn của toan máu do nhiễm toan chuyển hóa tăng clo máu, bởi vì chúng không có đủ anion tuần hoàn để chuyển hóa và sàn xuất ion bicarbonate, và toan máu nhanh chóng chuyển biến xấu đi khi truyền nhanh nước muối sinh lý, xem chi tiết ở phần trên.
Điều trị bằng NaHCO3 cũng có thể cân nhắc khi điều trị ban đầu trong nhóm nhỏ bệnh nhân được cho là tỷ lệ đào thài ceton acid thấp (tức là những bệnh nhân suy giảm nhận thức rõ rệt hoặc có rối loạn chức năng thận đang tiến triển từ trước [eGFR < 30 ml/ph]), để tránh giảm pH huyết tương 1 cách nguy hiểm và có thể gây xấu đi tình trạng huyết động. Đối với những bệnh nhân này, natri bicarbonate nên được truyền với tốc độ phù hợp với tốc độ sàn xuất ceton acid trong gan, khoảng 60 mmol/h, trên cơ sở dữ liệu từ một nghiên cứu ở bệnh nhân trưởng thành toan máu do nhịn đói. Dữ liệu về lợi ích của cách tiếp cận này đang thiếu trong thử nghiệm lâm sàng trong đó kết cục về phục hồi ổn định huyết động và tỷ lệ mắc các biến chứng như nhồi máu cơ tim, tổn thương thận cấp, đột quỵ được đánh giá trong nhóm nhỏ bệnh nhân trưởng thành bị toan máu mức độ trung bình – nặng (pH<7.2 và bicarbonate huyết tương <12 mmol/l) và trong một tình trạng huyết động không ổn định.
Trong nột nghiên cứu đa trung tâm, kiểm chứng bằng ca lâm sàng, hồi cứu trên bệnh nhi nhiễm toan ceton do đái tháo đường, Glaser cùng cộng sự đã quan sát thấy nguy cơ phù não gia tăng đáng kể ở những bệnh nhân có PaC02 thấp hoặc BUN cao khi có triệu chứng hoặc đã được truyền natri bicarbonate. Sự kết hợp này không chứng minh được nguyên nhân
cũng như không loại trừ các yếu tố gây nhiễu khác chưa được phân tích và có thể ảnh hưởng đến sự kết hợp này, đặc biệt liên quan sử dụng natri bicarbonate. Hơn thế nữa, nguy cơ phù não do truyền natri bicarbonate có thể gia tăng nếu bệnh nhân cũng nhận 1 lượng lớn insulin. Tuy nhiên, xét về tác hại có thể xảy ra, chúng tôi thống nhất rằng không nên truyền natri bicarbonate ở trẻ em nhiễm toan ceton do đái tháo đường trừ khi toan máu rõ rệt (pH <6.9 và nồng độ bicarbonate huyết tương < 5mmol/l) và chúng không có đáp ứng với thủ thuật tiêu chuẩn để khôi phục lại tình trạng huyết động.
Phù não trong quá trình điều tri
Tỷ lệ phù não trong quá trình điều trị nhiễm toan ceton do đái tháo đường ở trẻ em vẫn còn cao đến không chấp nhận được. Brown nhấn mạnh là biến chứng đáng sợ này xảy ra thường xuyên nhất lúc mới bắt đầu điều trị. Tại sao phù não lại xuất hiện khi bắt đầu điều trị? Có ý kiến cho rằng giảm tưới máu não đã xuất hiện trước khi điều trị nhiễm toan ceton đái tháo đường có thể gây khuynh hướng phù não lúc xảy ra tái tưới máu. Một nghiên cứu đối chứng ca lâm sàng về nhiễm toan ceton do đái tháo đường có biến chứng phù não ở trẻ em tại Anh cho thấy trẻ em diễn tiến phù não có mức độ toan máu nặng hơn những trẻ không tiến triển phù não. Trong nghiên cứu đó, cà 2 cách điều trị truyền insulin trong giờ đầu tiên sau khi bắt đầu điều trị và truyền lượng dịch lớn trong 4 giờ đầu (điều chỉnh theo độ nặng của toan máu) có liên quan tăng nguy cơ phù não.
Phù não xảy ra khi các tế bào trong não sưng lên, thể tích dịch ngoai bào tăng lên trong não hoặc cà 2. Các tế bào não phù lên khi có một lực thẩm thấu lớn tạo sự thuận lợi di chuyển nước trong tế bào, do tính thẩm thấu hiệu quà trong tế bào não cao hơn tính thẩm thấu hiệu quà trong huyết tương ở các mao mạch gần hàng rào máu – não. Các phương trình dưới đây cho thấy sự khác biệt giữa tính độ thẩm thấu huyết tương toàn phần và tính độ thẩm thấu huyết tương hiệu quà. Cà 2 cách tính đều dùng đơn vị mOsm/l. Để chuyển từ mg/dl sang mmol/l, nồng độ glucose máu trong huyết tương tính bằng đơn vị mg/dl nên chia cho 18 và nồng độ urea nitrogen máu (BUN) tính bằng mg/dl nên chia cho 2.8.
Trong cách tính độ thẩm thấu huyết tương toàn phần, người ta tính đến các thành phần thẩm thấu tham gia chính – Natri và các ion đi kèm cũng như glucose và ure. Do ure được vận chuyển qua hầu hết hàng rào tế bào và đạt được nồng độ bằng nhau trong dịch ngoại bào và dịch nội bào, nên ure không phải là chất thẩm thấu hiệu quả.
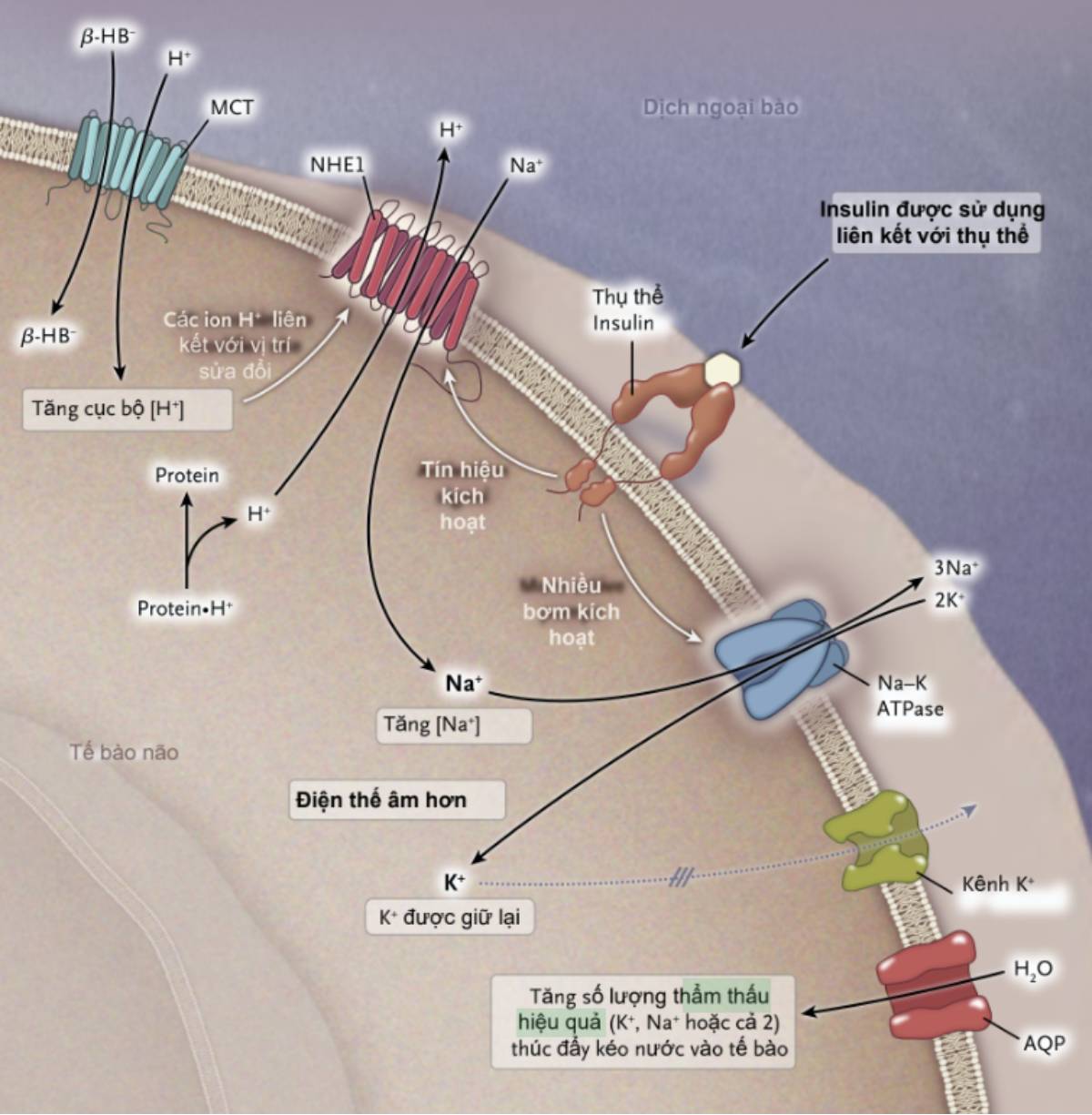
Tăng xâm nhập ^-hydroxybutyric acid vào trong tế bào thông qua chất đồng vận chuyển monocarboxylic acid (MCT) và sự phân ly sau đó thành các anion JỔ-HB– và H+ có thể dẫn đến tăng đáng kể nồng độ ion H+ trong khu vực màng tế bào cục bộ nơi tế bào thiết bị trao đổi natri – hydrogen 1 được đặt (NHE-1). NHE-1 được kích hoạt bởi nồng độ cao của H+ trong phần bên trong tế bào khi các ion H+ liên kết với một vị trí biến đổi NHE-1 sẽ làm tăng thẩm thấu hiệu quả các ion Na+ đi vào trong, trong khi đó phần lớn H+ thoát ra từ các tế bào này là thẩm thấu không hiệu quả; hầu hết các ion H+ này liên kết với các protein nội bào (protein«H+). Nếu dùng lượng lớn insulin trong quá trình điều trị, Na+ sẽ đi vào các tế bào này thông qua NHE-1 có thể được sản xuất nhờ Na-K-ATPase điện di, mà được insulin kích hoạt. Tăng điện thế âm nội bào có thể gây hiện tượng giữ lại các ion K+ trong những tế bào này, thúc đẩy dòng nước chảy qua thông qua các kênh nước aquaporin (AQP) trong các tế bào này. Cho dù quá trình này làm tăng Na+, K+ hoặc cả 2, thì hiệu quả cuối cùng là tăng số lượng thẩm thấu hiệu quả trong tế bào não.
Điều này nghĩa là ure không có khả năng tạo gradient thẩm thấu thông qua các hàng rào tế bào để ảnh hưởng đến sự phân bố nước giữa thể tích dịch ngoại bào và thể tích dịch nội bào, trong đó gồm các tế bào não. Do đó, ure nitrogen máu (BUN) trong công thức tính độ thầm thấu huyết tương hiệu quả ở bệnh nhân nhiễm toan ceton do đái tháo đường.
Tăng số lượng thẩm thấu não nội bào có thể xuất hiện cùng với tăng dòng natri đi vào các tế bào não (hình 3). Nồng độ cao các ion hydrogen trong tế bào não có thể kích hoạt các cơ chế vận chuyển ion natri trong màng tế bào, chủ yếu là cơ chế trao đổi natri – hydrogen. Nồng độ các ion hydrogen trong tế bào não có thể tăng khi acid beta-hydroxybutyric đi vào các tế bào trên cơ chế đồng vận chuyển monocarboxylic acid. Trao đổi cation này cũng được kích hoạt bởi nồng độ insulin trong dịch kẽ. Chúng tôi dự đoán sau khi bệnh nhân nhiễm toan máu nặng được truyền lượng lớn insulin, chất trao đổi natri-hydrogen 1 trong màng tế bào não có thể được kích hoạt. Điều này sẽ tăng số lượng thẩm thấu hiệu quà trong tế bào, do các ion natri đi vào chúng, trong khi phần lớn ion hydrogen thoát ra không hiệu quà về mặt thẩm thấu, do chúng liên kết với các protein nội bào. Truyền 1 liều insulin bolus là 1 phần điều trị ban đầu có thể mang lại tác dụng mạnh hơn trong việc kích hoạt chất trao đổi cation 1 này, khi hàng rào máu – não có thể dễ thấm insulin hơn. Các hướng dẫn hiện nay điều trị nhiễm toan ceton do đái tháo đường khuyến cáo không nên tiêm bolus insulin vào tĩnh mạch. Nếu trao đổi cation thông qua chất trao đổi ion natri – hydrogen 1 tăng hơn nữa khi pH trong dịch ngoại bào tăng, điều này có thể được giải thích ít nhất 1 phần là tăng nguy cơ phù não ở trẻ em nhiễm toan ceton do đái tháo đường khi truyền natri bicarbonate.
Giảm độ thẩm thấu hiệu quà trong huyết tương có thể do giảm nhanh nồng độ glucose huyết tương, tăng lượng nước không có chất điện giải hoặc cà 2. Giảm nhanh nồng độ glucose huyết tương xuất hiện khi lượng lớn glucose được chuyển hóa, bài tiết qua nước tiểu (do tăng GFR sau khi tái mở rộng thể tích máu động mạch hiệu quà), hoặc cà 2.
Tăng nước không có chất điện giải có thể do 1 số nguồn như dùng n engl j med 372; trong nước để ngăn ngừa tổn thương thần kinh do thiếu glucose não khi nồng độ glucose máu giảm. Sau khi chuyển hóa glucose, một lượng lớn nước không có chất điện giải có thể được giữ lại trong cơ thể. Các nguồn khác của nước không chứa chất điện giải không rõ ràng. Bệnh nhân nhiễm toan ceton do đái tháo đường thường tiêu thụ nhiều chất dịch chứa glucose (hoặc sucrose) hoặc nước để làm dịu cơn khát. Quá trình tiêu thụ dịch này có thể giữ lại trong dạ dày do tăng đường huyết làm chậm quá trình làm rỗng dạ dày. Nếu quá trình làm rỗng dạ dày xảy ra khi điều trị, dịch này sẽ được hấp thu trong ruột non và dẫn đến tăng lượng nước không có điện giải. Thực hành lâm sàng thường quy là truyền lượng lớn nước muối sinh lý cho bệnh nhân nhiễm toan ceton do đái tháo đường; nước không có điện giải có thể được tạo ra trong cơ thể thông qua khử muối. Nếu phóng thích vasopressin (do xuất hiện kích thích không thẩm thấu), tái hấp thu nước sẽ tăng trong các phần xa của nephron. Khi nồng độ glucose trong nước tiểu giảm, nồng độ natri sẽ tăng và lượng muối sinh lý dư thừa khi dùng sẽ thài qua nước tiểu dưới dạng dung dịch ưu trương (cho bệnh nhân)
Khoang kẽ của thể tích dịch ngoại bào trong não sẽ mở rộng nếu tăng áp suất thủy tĩnh mạo mạch, giảm áp suất thẩm thấu keo huyết tương, tăng tính thấm mao mạch hoặc tất cả yếu tố này. Do nước muối sinh lý được phân bố ban đầu trong huyết tương và tiến vào hàng rào máu- não trước khi cân băng với toàn bộ thể tích dịch ngoại bào, nên dùng quá nhiều nước muối sinh lý có thể làm tăng áp suất thủy tĩnh mao mạch, giảm nồng độ albumin máu và do đó giảm áp suất thẩm thấu keo mao mạch. Hơn thế nữa, có bằng chứng cho thấy hàng rào máu – não có thể bị rò rĩ ở thời điểm bệnh nhân nhiễm toan ceton được đưa vào bệnh viện.
Với phân tích như trên, một số biện pháp cần tập trung có thể cân nhắc khi điều trị bệnh nhân nhiễm toan ceton do đái tháo đường. Giảm nguy cơ phù não, chúng tôi đề nghị không được phép giảm độ thẩm thấu hiệu quà trong huyết tương trong 15 giờ điều trị đầu tiên, khoảng thời gian mà hầu hết xảy ra phù não. Đáng chú ý trong nghiên cứu của Glazer và cộng sự, không tăng nồng độ natri máu trong quá trình điều trị liên quan
tăng khả năng phù não. Khi K+ cần thiết, mục tiêu này có thể đạt được nếu Kali chloride thêm vào nước muối sinh lý 0.9%, với nồng độ 30-40 mmol/l. Cách điều trị này có độ thẩm thấu hiệu quà gần với nước tiểu ở những bệnh nhân này tại thời điểm đó. Do trẻ em nhiễm toan ceton đái tháo đường có nồng độ natri máu gần bình thường, mức độ tăng natri máu sẽ diễn tiến theo mức dịch truyền, nhưng đây là sự đánh đổi quan trọng để ngăn ngừa giảm độ thẩm thấu hiệu quà trong huyết tương. Nếu truyền glucose để ngăn ngừa tổn thương thần kinh do thiếu glucose não khi nồng độ glucose máu giảm, hãy thận trọng bù dịch sao cho thể tích nước không có chất điện giải là nhỏ nhất.
Bác sĩ lâm sàng nên khai thác kỹ tiền sử truyền dịch và xem các dấu hiệu lâm sàng chỉ ra dạ dày đang trống gần đây, với nguy cơ kèm theo là tái hấp thu nước không có chất điện giải ở ruột. Các dấu hiệu này là không giảm nhiều nồng độ glucose máu khi tăng bài tiết glucose qua nước tiểu hoặc đột ngột giảm độ thẩm thấu hiệu quà trong huyết tương, sẽ xảy ra nếu chỉ nhập nước mà không có đường. Hấp thu nhanh lượng nước uống vào có thể gây giảm đáng kể độ thẩm thấu hệu quà trong máu động mạch, hiện tượng này không thể phát hiện qua các xét nghiệm máu tĩnh mạch.
Chỉ nên truyền lượng lớn nước muối sinh lý chỉ khi cấp cứu huyết động. Mục tiêu truyền NaCl 0.9% nên là duy trì ổn định huyết động. Sử dụng Hct và Na+ máu để ước tính thể tích dịch ngoại bào và mức độ thiếu hụt ion natri trên từng bệnh nhân nhiễm toan ceton do đái tháo đường.
Tồng kết
Chúng tôi xem xét 3 vấn đề liên quan rối loạn kiềm toan và ý nghĩa lâm sàng các vấn đề này trên bệnh nhân nhiễm toan ceton do đái tháo đường. Toan máu trên hầu hết bệnh nhân có tình trạng này là do mất natri bicarbonate gián tiếp. Quá trình này không được gợi ý từ tỷ lệ 1:1 của sự tăng khoảng trống anion huyết tương đến giảm nồng độ bicarbonate huyết tương do cách tính này dựa vào “nồng độ” chứ không phải “hàm lượng”. Toan máu nặng trên một bệnh nhân nhiễm toan ceton do đái tháo đường có thể do giảm tốc độ loại bỏ ceton acid ở não và thận hơn là chỉ do gan tăng sàn xuất ceton acid quá mức. Kích hoạt chất trao đổi natri – hydrogen 1 trong các tế bào não do nhiễm toan ceton nội bào dẫn đến tăng độ thẩm thấu hiệu quà và do đó góp phần tiến triển phù não ở trẻ em nhiễm toan ceton do đái tháo đường.
Tài liệu tham khảo
- Emmett M. Anion-gap interpretation: the old and the new. Nat Clin Pract Nephrol 2006;2:4-5.
- Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol 2007;2: 162-74.
- Feldman M, Soni N, Dickson B. Influ- ence of hypoalbuminemia or hyperalbu- minemia on the serum anion gap. J Lab Clin Med 2005;146:317-20.
- Carvounis CP, Feinfeld DA. A simple estimate of the effect of the serum albu- min level on the anion Gap. Am J Nephrol 2000;20:369-72.
- Figge J, Jabor A, Kazda A, Fencl V. An- ion gap and hypoalbuminemia. Crit Care Med 1998;26:1807-10.
- Kamel KS, CheeiLêefa4ycrễta]HailassÈrìeaỉment. London: Taylor and Francisperin FA, Vasudevan S, Halperin ML. An- ion gap: may the anions restricted to the intravascular space undergo modification in their valence? Nephron 1996;73:382-9.
- Rastegar A. Use of the DeltaAG/Del- taHCO3- ratio in the diagnosis of mixed acid-base disorders. J Am Soc Nephrol 2007;18:2429-31.
- Goodkin DA, Krishna GG, Narins RG. The role of the anion gap in detecting and managing mixed metabolic acid-base disorders. Clin Endocrinol Metab 1984; 13:333-49.
- Adrogue HJ, Madias NE. Diabetic and other forms of ketoacidosis. In: Ge- nari FJ, Adrogue HJ, Galla JH, Madias NE, eds. Acid-base disorders and their 2005:313-51.
- Halperin ML, Kamel KS. Some obser- vations on the clinical approach to meta- bolic acidosis. J Am Soc Nephrol 2010;21: 894-7.
- Oh MS, Carroll HJ, Uribarri J. Mecha- nism of normochloremic and hyperchlo- remic acidosis in diabetic ketoacidosis. Nephron 1990;54:1-6.
- Gamblin GT, Ashburn RW, Kemp DG, Beuttel SC. Diabetic ketoacidosis present- ing with a normal anion gap. Am J Med 1986;80:758-60.
- Adrogué HJ, Wilson H, Boyd AE III, Suki WN, Eknoyan G. Plasma acid-base patterns in diabetic ketoacidosis. N Engl J Med 1982;307:1603-10.
- Narins RG, Cohen JJ. Bicarbonate therapy for organic acidosis: the case for its continued use. Ann Intern Med 1987; 106:615-8.
- Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001;38:703-27.
- Sabatini S, Kurtzman NA. Bicarbon- ate therapy in severe metabolic acidosis.J Am Soc Nephrol 2009;20:692-5.
- Kraut JA, Madias NE. Treatment of acute metabolic acidosis: a pathophysio- logic approach. Nat Rev Nephrol 2012;8: 589-601.
- Adrogué HJ, Madias NE. Management of life-threatening acid-base disorders — first of two parts. N Engl J Med 1998;338: 26-34.
- Mitchell JH, Wildenthal K, Johnson RL Jr. The effects of acid-base disturbanc- es on cardiovascular and pulmonary function. Kidney Int 1972;1:375-89.
- Wildenthal K, Mierzwiak DS, Myers RW, Mitchell JH. Effects of acute lactic acidosis on left ventricular performance. Am J Physiol 1968;214:1352-9.
- Davies AO. Rapid desensitization and uncoupling of human beta-adrenergic re- ceptors in an in vitro model of lactic aci- dosis. J Clin Endocrinol Metab 1984;59: 398-405.
- Huang YG, Wong KC, Yip WH, Mc- James SW, Pace NL. Cardiovascular re- sponses to graded doses of three cate- cholamines during lactic and hydrochloric acidosis in dogs. Br J Anaesth 1995;74: 583-90.
- Sonne O, Gliemann J, Linde S. Effect of pH on binding kinetics and biological effect of insulin in rat adipocytes. J Biol Chem 1981;256:6250-4.
- Gamba G, Oseguera J, Castrejón M, Gómez-Pérez FJ. Bicarbonate therapy in severe diabetic ketoacidosis: a double blind, randomized, placebo controlled trial. Rev Invest Clin 1991;43:234-8.
- Hale PJ, Crase J, Nattrass M. Meta- bolic effects of bicarbonate in the treat- ment of diabetic ketoacidosis. Br Med J (Clin Res Ed) 1984;289:1035-8.
- Morris LR, Murphy MB, Kitabchi AE. Bicarbonate therapy in severe diabetic ke- toacidosis. Ann Intern Med 1986;105:836- 40.
- Chua HR, Schneider A, Bellomo R. Bicarbonate in diabetic ketoacidosis — a systematic review. Ann Intensive Care 2011;1:23.
- Lipid metabolism: ketone bodies. In: Voet D, Voet JG. Biochemistry. 4th ed. New York: John Wiley, 2011:959-61.
- Nobes CD, Hay WW Jr, Brand MD. The mechanism of stimulation of respira- tion by fatty acids in isolated hepatocytes. J Biol Chem 1990;265:12910-5.
- Mitchell P. Coupling of phosphoryla- tion to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Na- ture 1961;191:144-8.
- Mitchell P. Keilin’s respiratory chain concept and its chemiosmotic conse- quences. Science 1979;206:1148-59.
- Flatt JP. On the maximal possible rate of ketogenesis. Diabetes 1972;21:50-3.
- Jungas RL, Halperin ML, Brosnan JT. Quantitative analysis of amino acid oxida- tion and related gluconeogenesis in hu- mans. Physiol Rev 1992;72:419-48.
- Kamel SK, Lin SH, Cheema-Dhadli S, Marliss EB, Halperin ML. Prolonged total fasting: a feast for the integrative physi- ologist. Kidney Int 1998;53:531-9.
- Owen OE, Felig P, Morgan AP, Wahren J, Cahill GF Jr. Liver and kidney metabo- lism during prolonged starvation. J Clin Invest 1969;48:574-83.
- Rolfe DF, Brand MD. The physiologi- cal significance of mitochondrial proton leak in animal cells and tissues. Biosci Rep 1997;17:9-16.
- Quant PA, Robin D, Robin P, Girard J, Brand MD. A top-down control analysis in isolated rat liver mitochondria: can the 3-hydroxy-3-methylglutaryl-CoA pathway be rate-controlling for ketogenesis? Bio- chim Biophys Acta 1993;1156:135-43.
- Owen OE, Morgan AP, Kemp HG, Sul- livan JM, Herrera MG, Cahill GF Jr. Brain metabolism during fasting. J Clin Invest 1967;46:1589-95.
- Brosnan JT, Lowry M, Vinay P, Gou- goux A, Halperin ML. Renal ammonium production — une vue canadienne. Can J Physiol Pharmacol 1987;65:489-98.
- Kety SS, Polis BD, Nadler CS, Schmidt CF. The blood flow and oxygen consump- tion of the human brain in diabetic acido- sis and coma. J Clin Invest 1948;27:500-10.
- Balaban RS, Mandel LJ. Coupling of aerobic metabolism to active ion transport in the kidney. J Physiol 1980;304:331-48.
- Gowrishankar M, Kamel KS, Hal- perin ML. A brain protein centered view of H+ buffering. J Am Soc Nephrol 2007; 18:2278-80.
- Savage MW, Dhatariya KK, Kilvert A, et al. Joint British Diabetes Societies guideline for the management of diabetic ketoacidosis. Diabet Med 2011;28:508-15.
- Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care 2009;32:1335-43.
- Glaser N, Barnett P, McCaslin I, et al. Risk factors for cerebral edema in chil- dren with diabetic ketoacidosis. N Engl J Med 2001;344:264-9.
- Wolfsdorf J, Craig ME, Daneman D, et al. Diabetic ketoacidosis in children and adolescents with diabetes. Pediatr Diabe- tes 2009;10:Suppl 12:118-33.
- Sperling MA. Cerebral edema in dia- betic ketoacidosis: an underestimated complication? Pediatr Diabetes 2006;7:73
- Brown TB. Cerebral oedema in child- hood diabetic ketoacidosis: is treatment a factor? Emerg Med J 2004;21:141-4.
- Glaser NS, Marcin JP, Wootton-Gorg- es SL, et al. Correlation of clinical and biochemical findings with diabetic keto- acidosis-related cerebral edema in chil- dren using magnetic resonance diffusion- weighted imaging. J Pediatr 2008;1 53: 541-6.
- Edge JA, Jakes RW, Roy Y, et al. The UK case-control study of cerebral oedema complicating diabetic ketoacidosis in children. Diabetologia 2006;49:2002-9.
- Dunger DB, Sperling MA, Acerini CL, et al. European Society for Paediatric En- docrinology/Lawson Wilkins Pediatric Endocrine Society consensus statement on diabetic ketoacidosis in children and adolescents. Pediatrics 2004;113(2):e133- e140.
- Hoorn EJ, Carlotti AP, Costa LA, et al. Preventing a drop in effective plasma os- molality to minimize the likelihood of cerebral edema during treatment of chil- dren with diabetic ketoacidosis. J Pediatr 2007;150:467-73.
- MacGregor IL, Gueller R, Watts HD, Meyer JH. The effect of acute hyperglyce- mia on gastric emptying in man. Gastro- enterology 1976;70:190-6.
- Carlotti AP, St George-Hyslop C, Guerguerian AM, Bohn D, Kamel KS, Hal- perin Ml. Occult risk factor for the devel- opment of cerebral edema in children with diabetic ketoacidosis: possible role for stomach emptying. Pediatr Diabetes 2009;10:522-33.
- Steele A, Gowrishankar M, Abraham- son S, Mazer CD, Feldman RD, Halperin ML. Postoperative hyponatremia despite near-isotonic saline infusion: a phenom- enon of desalination. Ann Intern Med 1997;126:20-5.
- Hoffman WH, Steinhart CM, el Gam- mal T, Steele S, Cuadrado AR, Morse PK. Cranial CT in children and adolescents with diabetic ketoacidosis. AJNR Am J Neuroradiol 1988;9:733-9.
- Krane EJ, Rockoff MA, Wallman JK, WolfsdorfJI. Subclinical brain swelling in children during treatment of diabetic ke- toacidosis. N Engl J Med 1985;312:1147- 51.
- Halperin ML, Maccari C, Kamel KS, Carlotti AP, Bohn D. Strategies to dimin- ish the danger of cerebral edema in a pe- diatric patient presenting with diabetic ketoacidosis. Pediatr Diabetes 2006;7:191
- Shafiee MA, Charest AF, Cheema- Dhadli S, et al. Defining conditions that lead to the retention of water: the impor- tance of the arterial sodium concentra- tion. Kidney Int 2005;67:613-21.
- Napolova O, Urbach S, Davids MR, Halperin ML. Assessing the degree of ex- tracellular fluid volume contraction in a patient with a severe degree of hypergly- caemia. Nephrol Dial Transplant 2003;18: 2674-7.








{kind=link}